
The link between inner ear malformations and the rest of the body: what we know so far about genetic, imaging and histology:
Editorial
Published: 03 March 2020
The link between inner ear malformations and the rest of the body: what we know so far about genetic, imaging and histology
Felice D’Arco,
Eser Sanverdi,
William T. O’Brien,
Ajay Taranath,
Giacomo Talenti &
Susan I. Blaser
Neuroradiology (2020)Cite this article
Metricsdetails
The complexity of ear development
Introduction
The inner ear has complex anatomy with several genes involved in its development. A spectrum of malformations has been described with different proposed pathogenesis, clinical implications and associated sensorineural hearing loss (SNHL) [1].
Hereditary hearing loss (HL) encompasses approximately 50% of congenital SNHL cases and syndromic associations have been described with additional abnormalities involving other organ systems [2]. These syndromes may have genetic and/or environmental causes, posing interesting questions on the developmental and genetic links between the ear and the rest of the body [3]. There has been an explosion in the identification of genetic mutations causing SNHL: whole exome and whole genome sequencing currently takes weeks to months and remains costly; on the other hand, phenotype-genotype prediction based on imaging studies may help guide the clinician towards a more targeted gene panel.
Advances in knowledge on the embryologic development of inner ear structures
Until recently, the window for insults that can be responsible for inner ear malformations was thought to be limited to the period between the 3rd and the 7th gestational weeks [4]. However, more recent updates on inner development suggest that the cochlea completes one turn by 8 weeks gestation, two turns by 10 weeks gestation and the final half turn is not completed until 25 weeks gestation [4]. Thus, the window for an insult seems to be wider and specifically the period between 8 and 10 weeks gestation is probably critical in the setting of cochlear hypoplasias, which are more frequent and complex entities than previously thought [1, 4]. Furthermore, the completely formed membranous labyrinth continues to grow until midterm, when the cartilaginous matrix surrounding the membranous matrix undergoes ossification between 16 and 23 weeks gestation; this process is impaired in the incompletely partitioned cochleas (IP). Finally, the vestibular aqueduct, which is enlarged in several genetic causes of SNHL, continues to develop after birth until puberty [4]. Therefore, the window for inner ear maldevelopment extends beyond early gestation.
Current understanding of mechanisms of inner ear malformations
In terms of inner ear malformations, it was previously thought that insults (vascular, infectious or toxic) during membranous labyrinthine development were the primary cause of inner ear malformations. The thalidomide model of fibrin emboli and focal hypoperfusion is a good example of ototoxic embryopathy [5]; however, the possibility of a genetic vascular component of this model has recently been raised, as HOXA1 mutations may be associated with a similar clinical phenotype characterized by abnormalities of the cerebrovascular system and the inner ears. [6]. This is a good example of how genetic and toxic mechanisms can be both implied in the pathogenesis of congenital deafness.
If the development of the temporal bone and inner ear structures is more complex than previously thought [4], the pathogenesis of inner ear malformations is probably more complex as well. Several important gene families, such as FOX, DIX, FGF and SOX, are critical in the embryogenesis of the otic capsule, and associations have been found between specific genes and characteristic inner ear malformations. For example, in murine models, loss of Pax2 results in isolated cochlear aplasia, loss of Dlx5 is responsible for absence of posterior and superior semicircular canals, loss of Otx1 is associated to absence of the lateral semicircular canal, and Nkx5-1 is instrumental in the development of all semicircular canals [7, 8].
Moreover, most genes involved in inner ear development also play important roles in the embryogenesis of other systems. For example, a recently published paper described the critical role of FOXI1 in both maintaining the correct fluid composition of the labyrinth, and maintaining homeostasis of electrolytes within the renal ductal system. Recessive loss of function of FOXI1 results in distal renal tubular acidosis and SNHL associated with bilateral and symmetric vestibular aqueduct dilatation [9].
In light of recent literature, including that regarding whole exome and next-generation sequencing in SNHL, it is clear that there is more to the picture than meets the eye when it comes to the embryology and pathogenesis of inner ear development and malformations. Radiologists need to be aware of these newer evidences, to better understand these malformations and their secondary associations. A brief discussion of two categories of inner ear dysplasia (cochlear hypoplasia and incomplete partition) and their syndromic or “other organ” associations follows.
Cochlear hypoplasia
Cochlear hypoplasias (CHs) account for 15–23% of congenital inner ear abnormalities and are categorized as types 1–4. The internal cochlear architecture may be normal or abnormal depending on the subtype. A small cochlea with absent or incomplete internal structure is categorized as types 1 (bud-like) and 2 (cystic hypoplastic cochlea), respectively. Types 3 (fewer than 2 turns and/or small turns with short modiolus) and 4 (hypoplastic middle and apical turns) present with relatively preserved internal cochlear partitioning with the difference being that type 4 has a preserved basal turn and very small and “flattened” upper turns [1, 10]. In other words, CH types 1 and 2 are small versions of an incompletely partitioned cochlea, while CH types 3 and 4 are small cochleas but with almost complete internal structure.
Despite this distinction in 4 groups is clinically useful, in our experience, CHs are more a continuum of malformations, differently from the 3 types of incompletely partitioned cochleas that are more distinct groups in terms of morphology and etiopathogenesis (see below).
A specific type of CH in association with non-inner ear anomalies may help predict the appropriate genetic pathway to interrogate
Syndromic cases of CH often have associated abnormalities in other organ systems that should be sought when characteristic inner ear malformations are identified, and vice versa (Fig. 1). In fact, there are some CH disorders with fairly specific inner ear imaging phenotypes predicting the genotype.
Fig. 1
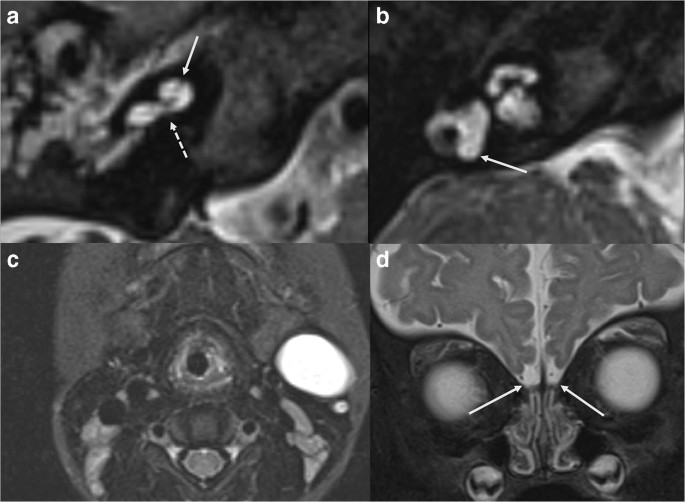
Axial 3D T2 WI showing two types of cochlear hypoplasia in syndromic causes of hearing loss. a Typical cochlear appearances in BOR syndrome: unwound cochlea with offset of the hypoplastic middle and apical turns (arrow) away from a tapered basal turn (dashed arrow). b Cochlear hypoplasia in a patient with SOX10 mutation: flattened cochlea with all turns and interscalar septations present but small, fitting the definition of CH type 3. Note the absent posterior semicircular canal (arrow) and small lateral semicircular canal. c Axial T2 WI STIR of the neck showing a 2nd branchial cleft anomaly in a patient with BOR syndrome (same subject of image a). d Bilateral olfactory bulb hypoplasia (arrows) in a patient with SOX10 mutation (same subject of image b)
Full size image
Inner ear imaging in CHARGE syndrome (CHD7 mutation) is so specific that the presence of cochlear hypoplasia and bony cochlear nerve canal (BCNC) hypoplasia (or “cochlear aperture stenosis”) in association with typical absence of the semicircular canals is very suggestive of CHARGE syndrome [11]. The Verloes’ criteria include major (coloboma of the eye(s), choanal atresia, and hypoplastic semicircular canals) and minor findings, including brainstem and cranial nerve dysfunction, growth hormone and gonadotropin deficiencies, abnormal middle or external ear and malformation of mediastinal organs [12] and represent a good example of association between inner ear and other organ system malformations. Interestingly, CHARGE may also have hypoplastic/aplastic olfactory apparatus and hypogonadism. In patient with hypoplastic/aplastic olfactory apparatus (i.e. Kallmann phenotype (KS)), most likely mutations are in KAL1 or FGFR1 gene; however, KS with hearing loss is extremely unusual and should prompt testing for the less common CHD7 (CHARGE) mutations and also SOX10 (Waardenburg syndrome) mutations [12, 13].
Imaging of inner ears in branchio-oto-renal BOR (EYA1, SIX1 and SIX5 mutations) is also typical, demonstrating an unwound appearing cochlea with offset of the hypoplastic middle and apical turns away from a tapered basal turn (Fig. 1a). The labyrinthine segment of the facial nerve canal exits from the IAC early and middle ear ossicles are typically malformed. Associated non-labyrinthine anomalies in BOR include branchial cleft cysts and fistulae (Fig. 1c) and renal anomalies [14].
In others, it is the combination of petrous and non-labyrinthine features that help predict the genetic mutation. As an example, in Waardenburg syndrome–associated deafness (SOX10, PAX3, MITF, EDN3, EDNRB, SNA12 mutations), there is often a hypoplastic flattened cochlea, although all turns and interscalar septations are present (CH3) and there are characteristic abnormalities of the semicircular canals, such as persistent anlage of the posterior semicircular canal (Fig. 1b) [15]. Waardenburg syndrome is considered a neurocristopathy and variable non-inner ear features may include white matter signal abnormalities, Hirschsprung intestinal aganglionosis, white forelock, depigmentation of skin patches, intensely blue eyes or iris heterochromia and dystopia canthorum, agenesis or hypoplasia of the olfactory bulbs and nerves, isolated cerebellar dysplasia and parotid and lacrimal gland absence or hypoplasia [2]. Among different types of Waardenburg, SOX10 mutation is characterized by almost pathognomonic combination of inner ear malformations as described above, Kallmann syndrome and Hirschsprung disease (Fig. 1d) [2].
Down syndrome (trisomy 21) patients have measurable hypoplasia of the cochlea, cochlear nerve canal stenosis and of the small bony island of the semicircular canals associated with characteristic facial abnormalities [2].
Incomplete partition
Cochlear incomplete partition (IP) is divided into 3 groups based upon the pattern of abnormal internal structure. Unlike the lack of cochlea-vestibular separation present in the common cavity, in the IP category, the cochlea is distinguishable from the vestibule [16] (Fig. 2). The underlying pathogenesis of IP anomalies varies depending on the subtype.
Fig. 2
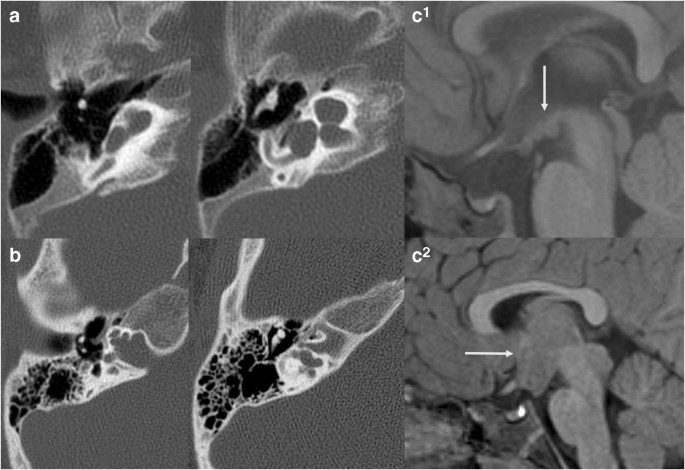
a IP1 in split-hand-foot-malformation (SHFM1) patient: Axial CT images demonstrate typical features of IP1 including dilated basal turn and feature-less cochlea. This malformation has been mistakenly called Mondini malformation (IP2); however, the basal turn is normal in size in IP2 and dilated in IP1. DLX5 is one of the genes implicated in SHFM. b Axial CT images showing typical features of IP3 in a male with X-linked deafness due to POU3F4 mutation. There is typical widening of the fundus of the IAC and absence of the lamina cribrosa, the bony plate between the fundus and the cochlea. The modiolus is not identified. There is also medialization of the facial nerve canal. c Sagittal T1 WI in the midline of the brain in 2 patients with IP3 and X-linked deafness showing hypothalamic malformations ranging from subtle deformity (upper row) to large hamartoma-like mass (lower row)
Full size image
(1)
IP1 is characterized by near-complete absence of the modiolus, interscalar septum (with shallow profile of the cochlea) and lamina spiralis (Fig. 2a). There is no unifying genetic aetiology known at this time. It has been postulated that vascular supply from the internal auditory canal (IAC) is necessary for appropriate development of internal cochlear structure and that defective blood flow may be the cause of this malformation [1].
(2)
IP2 is characterized by deficient internal structures limited to the upper part of the cochlea. The basal turn is normal and there is variable absence of the upper portion of the interscalar septum in the lateral aspect of the cochlea, in association with enlarged vestibular aqueduct (VA) and dilated vestibule (so-called Mondini triad). Mondini triad is the radiological hallmark of SLC26A mutations (Pendred syndrome); the associated non-labyrinthine finding in the Pendred syndrome is thyroid goitre secondary to iodine organification defect [2]. Dilated vestibular aqueducts may also be seen in isolation with KCNJ10 and FOXI1 mutations [17]. Transmission of high CSF pressure into the cochlea from the enlarged VA has been advocated as a possible mechanism for the dilatation of the upper part of the cochlea and widening of the scala vestibuli [1]; however, the stability overtime of cochlear enlargement on imaging, the different embryological origin of the distal and proximal interscalar septum [4], the fact that on histology the endolymphatic duct and sac are not surrounded by a conspicuous perilymphatic space [18], and the genetic link between development of the cochlea and vestibular aqueduct [3] are not supportive of this hypothesis in our opinion.
(3)
IP3 is the radiological hallmark of X-linked deafness due to POU3F4 mutations (and rarely COL4A6 mutations, which are radiologically indistinguishable) [1, 19]. In animal models, pou3f4 is expressed in the otic mesenchymal cells and regulate spiral ganglion axon fasciculation [20]. In humans, IP3 is characterized by a large IAC with a dilated fundus, absent modiolus and absent lamina cribrosa separating the cochlea (perilymph) from the internal auditory canal (subarachnoid space). Bony interscalar septa are relatively preserved, giving specific corkscrew shape of the cochlea (Fig. 2b). The facial nerve canal is proximally located and may be dilated. Ossicular anomalies include fixed stapes footplate in most and stapedectomy leads to a perilymph gusher. Since the outer periosteal and middle endochondral layers of the otic capsule receive vascular supply from the middle ear mucosa, and these layers seem to be abnormally thin in IP3, it is possible that deficient vascular supply from middle ear vessels is involved in the pathogenesis of this malformation [1]. Interestingly, most of these patients have associated dysmorphisms of the hypothalamus [21], and POU3F4 plays a role in the development of the brain, particularly the hypothalamic nuclei (Fig. 2c). This implies a developmental link between the hypothalamus and inner ear, which is confirmed in reported cases of Pallister-Hall syndrome, characterized by large hamartoma-like lesions and cochlear hypoplasia [21].
Another intriguing link between brain and inner ear development is the association between inner ear malformations and holoprosencephaly (Fig. 3). Syntelencephaly, or middle interhemispheric variant, is a subtype of holoprosencephaly, characterized by an abnormal connection of the cerebral hemispheres across midline. Mutation of ZIC2 gene has been implicated in the pathogenesis of this particular brain malformation, and the same Zic2 gene has been linked in animal models to the development of the otic capsule [22], explaining the coexistence of inner ear malformations in patients with holoprosencephaly and holoprosencephaly variants.
Fig. 3
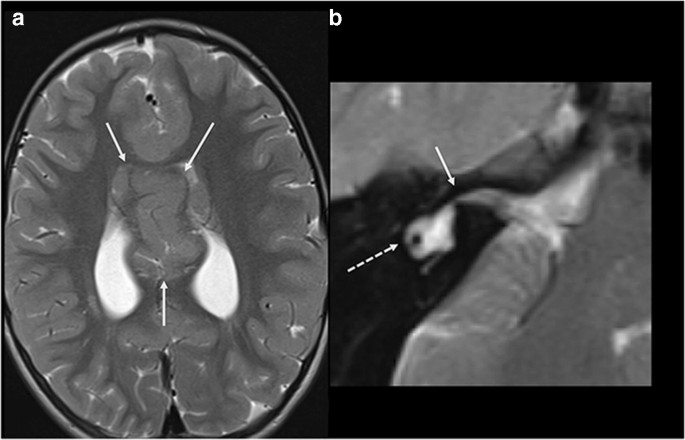
a Axial T2 WI of the brain in a patient with syntelencephaly (arrows). b Axial 3D T2 WI at the level of the inner ear in the same patient shows associated right cochlear aplasia (arrow) and dysplastic lateral semicircular canal (dashed arrow)
Full size image
Conclusion
A bulk of new evidences regarding factors associated with inner ear development and malformations is on the rise, greatly improving our understanding of these complex anomalies. In particular, portions of inner ear structures continue to develop beyond the early gestational period, contrary to what was previously thought. In addition, numerous gene mutations have been found in association with inner ear malformations, many of which affect other organ systems as well. The classification of cochlear malformations, including four subtypes of cochlear hypoplasia and three subtypes of incomplete partition, should motivate radiologists to look for diagnostic phenotype-genotype correlations, as well as associated malformations in other organ systems. Syndromic causes of hearing loss, in particular, may often be characterized by typical imaging and phenotypic findings within and outside the temporal bone. “Connecting the dots” between development and malformations of the ear, brain and other organ systems looks to be a promising endeavour for radiologists in the genomics era.
References
1.
Sennaroglu L (2016) Histopathology of inner ear malformations: do we have enough evidence to explain pathophysiology? Cochlear Implants Int 17:3–20. https://doi.org/10.1179/1754762815Y.0000000016
Article
PubMed
Google Scholar
2.
D’Arco F, Youssef A, Ioannidou E, et al. (2019) Temporal bone and intracranial abnormalities in syndromic causes of hearing loss: an updated guide, Eur J Radiol. 26;123:108803. doi: https://doi.org/10.1016/j.ejrad.2019.108803
Article
Google Scholar
3.
Koffler T, Ushakov K, Avraham KB (2015) Genetics of hearing loss: syndromic. Otolaryngol Clin N Am 48:1041–1061. https://doi.org/10.1016/j.otc.2015.07.007
Article
Google Scholar
4.
P.M. Som, H.D. Curtin, K. Liu et al. (2016) Current embryology of the temporal bone, part I: the inner ear. Neurographics 6(4):250 –265; doi:https://doi.org/10.3174/ng.4160166
Article
Google Scholar
5.
Parsa CF, Robert MP (2013) Thromboembolism and congenital malformations: from Duane syndrome to thalidomide embryopathy. JAMA Ophthalmol 131(4):439–447
CAS
Article
Google Scholar
6.
Tischfield MA, Bosley TM, Salih MAM, Alorainy IA, Sener EC, Nester MJ, Oystreck DT, Chan WM, Andrews C, Erickson RP, Engle EC (2005) Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet 37:1035–1037. https://doi.org/10.1038/ng1636
CAS
Article
PubMed
Google Scholar
7.
Burton Q, Cole LK, Muhleisen M et al (2004) The role of Pax2 in mouse inner ear development. Dev Biol 272:161–175. https://doi.org/10.1016/j.ydbio.2004.04.024
CAS
Article
PubMed
Google Scholar
8.
Hadrys T, Braun T, Rinkwitz-Brandt S, Arnold HH, Bober E (1998) Nkx5-1 controls semicircular canal formation in the mouse inner ear. Development 125:33–39
CAS
PubMed
Google Scholar
9.
Enerbäck S, Nilsson D, Edwards N, Heglind M, Alkanderi S, Ashton E, Deeb A, Kokash FEB, Bakhsh ARA, van’t Hoff W, Walsh SB, D’Arco F, Daryadel A, Bourgeois S, Wagner CA, Kleta R, Bockenhauer D, Sayer JA (2018) Acidosis and deafness in patients with recessive mutations in FOXI1. J Am Soc Nephrol 29:1041–1048. https://doi.org/10.1681/ASN.2017080840
Article
PubMed
Google Scholar
10.
Talenti G, Manara R, Brotto D, D’Arco F (2018) High-resolution 3 T magnetic resonance findings in cochlear hypoplasias and incomplete partition anomalies: a pictorial essay. Br J Radiol 91:20180120. https://doi.org/10.1259/bjr.20180120
Article
PubMed
Google Scholar
11.
A.K. Morimoto A.K, Wiggins III R.H, Hudgins P.A, et al. (2006) Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings, AJNR Am J Neuroradiol 27:1663–1671
12.
Verloes A (2005) Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet 133A:306–308
Article
Google Scholar
13.
Pingault V, Bodereau V, Baral V, Marcos S, Watanabe Y, Chaoui A, Fouveaut C, Leroy C, Vérier-Mine O, Francannet C, Dupin-Deguine D, Archambeaud F, Kurtz FJ, Young J, Bertherat J, Marlin S, Goossens M, Hardelin JP, Dodé C, Bondurand N (2013) Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet 92:707–724. https://doi.org/10.1016/j.ajhg.2013.03.024
CAS
Article
PubMed
PubMed Central
Google Scholar
14.
Hsu A, Desai N, Paldino MJ (2018) The unwound cochlea: a specific imaging marker of branchio-oto-renal syndrome. AJNR Am J Neuroradiol. https://doi.org/10.3174/ajnr.A5856
CAS
Article
Google Scholar
15.
Elmaleh-Bergès M, Baumann C, Noël-Pétroff N et al (2013) Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations. AJNR Am J Neuroradiol 34:1257–1263. https://doi.org/10.3174/ajnr.A3367
Article
PubMed
Google Scholar
16.
Sennaroglu L, Saatci I (2002) A new classification for cochleovestibular malformations. Laryngoscope 112:2230–2241. https://doi.org/10.1097/00005537-200212000-00019
Article
PubMed
Google Scholar
17.
Liu Y, Wang L, Feng Y et al. (2016) A new genetic diagnostic for enlarged vestibular aqueduct based on next-generation sequencing PLoS One. Dec 20;11(12):e0168508. doi: https://doi.org/10.1371/journal.pone.0168508
Article
Google Scholar
18.
Lo WW, Daniels DL, Chakeres DW, Linthicum FH Jr, Ulmer JL, Mark LP, Swartz JD (1997) The endolymphatic duct and sac. AJNR Am J Neuroradiol 18:881–887
CAS
PubMed
Google Scholar
19.
Rost S, Bach E, Neuner C, Nanda I, Dysek S, Bittner RE, Keller A, Bartsch O, Mlynski R, Haaf T, Müller CR, Kunstmann E (2014) Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur J Hum Genet 22:208–215
CAS
Article
Google Scholar
20.
Coate TM, Raft S, Zhao X et al (2012) Otic mesenchyme cells regulate spiral ganglion axon fasciculation through a Pou3f4/EphA4 signaling pathway. Neuron 73:49–63. https://doi.org/10.1016/j.neuron.2011.10.029
CAS
Article
PubMed
PubMed Central
Google Scholar
21.
Siddiqui A, D’Amico A, Colafati GS, Cicala D, Talenti G, Rajput K, Pinelli L, D’Arco F (2019) Hypothalamic malformations in patients with X-linked deafness and incomplete partition type 3. Neuroradiology 61:949–952. https://doi.org/10.1007/s00234-019-02230-z
Article
PubMed
Google Scholar
22.
Chervenak AP, Bank LM, Thomsen N, et al. (2014) The role of Zic genes in inner ear development in the mouse: exploring mutant mouse phenotypes. Dev Dyn 243:1487– 1498. doi: https://doi.org/10.1002/dvdy.24186
CAS
Article
Google Scholar
Download references
Funding
No funding was received for this study.
Author information
Author notes
FD’A and ES are joint first authors and contributed equally to this work.
Affiliations
Department of Radiology, Great Ormond Street Hospital for Children, London, UK
Felice D’Arco
& Eser Sanverdi
Department of Radiology and Medical Imaging, Cincinnati Children’s Hospital Medical Center, Cincinnati, USA
William T. O’Brien
Department of Radiology, Women’s and Children’s Hospital, North Adelaide, South Australia, Australia
Ajay Taranath
Department of Diagnostics and Pathology, Neuroradiology Unit, Verona University Hospital, Verona, Italy
Giacomo Talenti
Division of Neuroradiology, Department of Diagnostic Imaging, The Hospital for Sick Children, Toronto, Ontario, Canada
Susan I. Blaser
Corresponding author
Correspondence to Felice D’Arco.
Ethics declarations
Conflict of interest
None identified.
Ethical approval
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
NA
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Reprints and Permissions
About this article

Cite this article
D’Arco, F., Sanverdi, E., O’Brien, W.T. et al. The link between inner ear malformations and the rest of the body: what we know so far about genetic, imaging and histology. Neuroradiology (2020). https://doi.org/10.1007/s00234-020-02382-3
Download citation
Received26 November 2019
Accepted17 February 2020
Published03 March 2020
DOIhttps://doi.org/10.1007/s00234-020-02382-3
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου