
Casos Clínicos,
PLASMOCITOMA EXTRAMEDULAR NASOFARINGEO: INFORME DE UN CASO
El plasmocitoma extramedular de nasofaringe es una neoplasia de células plasmáticas inusual, que comprende un 4% de las neoplasias no epiteliales a este nivel. Aparece como una lesión única, de color rojo violáceo, en varones mayores de 40 años y puede ocasionar epistaxis.
Coautores
Rosario Guerrero Cauqui* Carmen Benítez García** Regla Gallego Gallegos***
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras*
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras, Algeciras, España**
Doctora, Hospital de Algeciras, Algeciras, España***
Rosario Guerrero Cauqui* Carmen Benítez García** Regla Gallego Gallegos***
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras*
Doctora Especialista en Anatomía Patológica, Hospital de Algeciras, Algeciras, España**
Doctora, Hospital de Algeciras, Algeciras, España***
Clasificación en siicsalud
Artículos originales > Expertos del Mundo >
página /dato/casiic.php/156201
Especialidades







Artículos originales > Expertos del Mundo >
página /dato/casiic.php/156201
Especialidades
Primera edición en siicsalud
1 de diciembre, 2017
1 de diciembre, 2017
PLASMOCITOMA EXTRAMEDULAR NASOFARINGEO: INFORME DE UN CASO
(especial para SIIC © Derechos reservados)
(especial para SIIC © Derechos reservados)
Introducción
Las neoplasias de células plasmáticas incluyen tres entidades diferentes: mieloma múltiple, enfermedad diseminada (la más frecuente, 1.5% de todas las neoplasias del organismo), y dos formas localizadas, plasmocitoma solitario, con médula ósea y esqueleto normales (según las guías clínicas del International Myeloma Working Group de 2013): plasmocitoma medular que surge en hueso (vértebras, fémur y pelvis), llamado plasmocitoma solitario óseo, y otro en tejidos blandos o plasmocitoma solitario extramedular.1,2
Caso clínico Mujer de 65 años, hipertensa, que acude al Servicio de Urgencias por un episodio de epistaxis autolimitada en FND, coincidiendo con un catarro.
Se le hace una rinofibrolaringoscopia y se aprecia una lesión levemente rojiza de mucosa de aspecto normal en zona anterior de nasofaringe y rodete tubárico derecho. Se le hace una RM que muestra masa lobulada de 13 x 11 mm en mucosa anterior derecha de nasofaringe, que no se realza tras contraste ni infiltra estructuras y sin adenomegalias (Figura 1). Se informa al paciente de la necesidad de realizar una biopsia y su posible exéresis, y ésta acepta, realizándose su extirpación completa sin complicaciones mediante CENS.

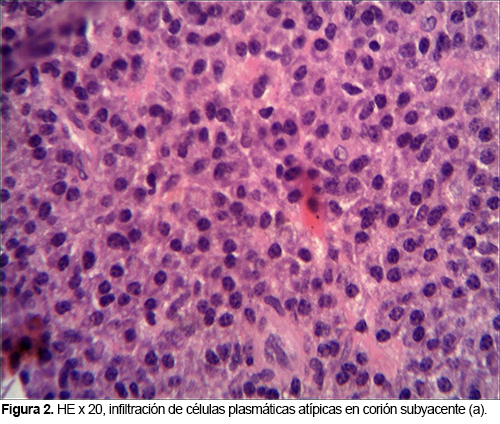
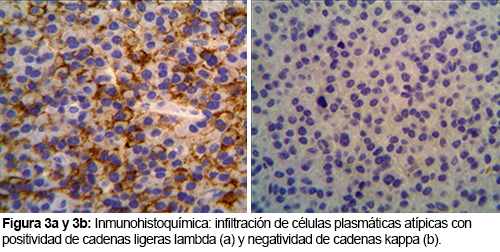
El estudio histológico dio como resultado la presencia de un plasmocitoma (Figuras 2 y 3): infiltrado subepitelial por células plasmáticas con atipia que muestra fenotipo CD138+CD56+/- CD10 con restricción de cadenas ligeras kappa.
Se remite al Servicio de Hematología para completar su estudio, que solicita bioquímica, proteinograma, células plasmáticas y gammagrafía ósea que resultan normales. La cuantificación de inmunoglobulinas es normal, IF y CLLs son negativos, la punción aspirativa de médula ósea no detecta infiltración por células plasmáticas patológicas ni por citometría; y la RM cervical muestra signos de espondiloartrosis, osteocondrosis con osteofitos, y cambios degenerativos.
La paciente no presentó en ningún momento manifestaciones clínicas, y ante los resultados obtenidos, se decide someterla a revisiones periódicas. Luego de cinco años sigue asintomática, sin signos de recidiva locorregional.
Discusión
El plasmocitoma extramedular es una proliferación de células plasmáticas, considerada un linfoma de bajo grado de malignidad en la clasificación de Kiel, caracterizada por expansión monoclonal de células plasmáticas productoras de cadenas ligeras de inmunoglobulinas. Constituye el 3% de los tumores de células plasmáticas. Pueden asentarse en cualquier zona del organismo que contenga tejido linfoide (ganglios linfáticos, piel, tracto aerodigestivo), aunque se localiza preferentemente (80%) en submucosa del tracto aerodigestivo superior: en cavidad nasal (46%) senos paranasales (36%) orofaringe (25%), nasofaringe (20%), laringe (4%) y glándula tiroidea.1 Fue descrito por Schraddle en 1905, y posteriormente por Ewing. Su incidencia se estima en un caso por 500 000 personas.
Son tumores infrecuentes, que representan menos del 1% (0.4%) de las neoplasias de cabeza y cuello, y menos del 0.5% de las neoplasias malignas del tracto respiratorio superior, siendo el 4% del total de los tumores no epiteliales de cavidad nasal y senos paranasales.2
Se presenta en adultos mayores de 40 años, entre la quinta y séptima década, sobre todo en los hombres en una proporción 3:1, especialmente en los de origen caucásico. Se ha relacionado con diversos factores como irritación crónica por sustancias inhaladas e infecciones por virus, aunque no está clara su etiología. La clínica es inespecífica, dependiendo de la localización y el tamaño: tumor o edema local (80%), obstrucción nasal crónica (35%), epistaxis (35%), dolor localizado (20%), proptosis (15%), rinorrea (10%), cefalea. Raramente se presenta con disminución de la visión (parálisis del VI par craneal, proptosis, pólipo nasal, y adenopatías. Suele surgir simultáneamente en varias zonas (20%).2,3
Macroscópicamente, aparece como una masa circunscripta a submucosa de consistencia firme, no ulcerada, sésil o pedunculada, de coloración variable, sobre todo de un rojo violáceo, fácilmente sangrante. Histológicamente no se distingue del mieloma, es una proliferación difusa de plasmocitos maduros; aunque no se puede confirmar que sea una lesión precursora del mieloma.4 Al ser un tumor submucoso, deberían realizarse biopsias profundas, para que su resultado no sea negativo.
A nivel microscópico muestra una morfología blastoide con presencia de inmunoblastos, pero con inmunofenotipo de diferenciación plasmocitaria (EMA+, CD138+, CD38+, CD56+). Puede producir inmunoglobulinas, sobre todo IgG (55%) e IgA (21%). Mediante inmunohistoquímica se pone de manifiesto la producción de cadenas pesadas IgG e IgA, y cadenas ligeras kappa o lambda.5
Su diagnóstico se realiza con el estudio histológico y de extensión sistémico negativo, estudio de sangre periférica, proteinograma, proteinuria, inmunoelectroforesis sérica y urinaria, gammagrafia ósea y punción de médula ósea. Las pruebas TAC y RM son útiles para establecer la extensión de la enfermedad, careciendo de signos propios para el diagnóstico. Los criterios clínicos para su diagnóstico son: confirmación histológica, presencia de uno o dos focos de enfermedad, biopsia de médula ósea normal, radiología ósea normal, ausencia de anemia, hipercalcemia e hiperuricemia; la presencia de proteínas monoclonales en orina o suero no excluye el diagnóstico.6.7 Si se localiza en la cavidad nasal, se debe hacer diagnóstico diferencial con tumores hemorrágicos, sobre todo carcinoma escamoso, y otros como melanoma, carcinoma indiferenciado, linfoma, adenoma pituitario, entre otros.1,8
Existe controversia respecto de su evolución y tratamiento, atribuible a su baja incidencia. Generalmente, tienen una progresión locorregional: enfermedad solitaria en aproximadamente 40%, diseminación linfática del 10% al 21%, segunda localización (ósea en 38%, tejidos blandos en 20%, y ambas en un 42%, y raramente se diseminan a mieloma múltiple, incluso después de muchos años, a diferencia del plasmocitoma medular. Entre los factores que se relacionan con progresión a mieloma está se destacan la edad mayor de 60 años, el tamaño del tumor y la persistencia de proteína M luego del tratamiento.8,9
El tratamiento está cuestionado, pues se considera de elección la radioterapia, al ser altamente sensible, con una alta tasa de control local (mayor del 80%), se recomiendan utilizarla incluso después de la extirpación macroscópica total, dependiendo de la localización, a dosis de al menos 40 Gy en cuatro semanas, y si es mayor de 5 cm, dosis por encima de 50 Gy. Algunos autores aconsejan incluir todo el hueso de campo de radiación, debido a las recurrencias.10,11 Otros aconsejan la escisión de la lesión con márgenes libres por vía endonasal (CENS), siempre que el tumor sea accesible.2,9 Otros autores encuentran mejores resultados en la modalidad combinada de cirugía más radioterapia. Si los ganglios son positivos, se deben incluir en el campo a irradiar, de 40 Gy, o vaciamiento cervical seguido de RT.9-11
Conclusiones
El plasmocitoma extramedular es un linfoma de células plasmáticas de bajo grado de malignidad e infrecuente, que no afecta la médula ósea ni el tejido óseo, y aparece habitualmente en varones mayores de 40 años, como una masa de color rojo violácea no ulcerada. Puede manifestarse como epistaxis unilateral, por lo que debería realizarse una fibrolaringoscopia, como protocolo en todas las epistaxis unilaterales no recidivantes, independientemente de la edad, para descartar tumores a nivel de la rinofaringe.
Las neoplasias de células plasmáticas incluyen tres entidades diferentes: mieloma múltiple, enfermedad diseminada (la más frecuente, 1.5% de todas las neoplasias del organismo), y dos formas localizadas, plasmocitoma solitario, con médula ósea y esqueleto normales (según las guías clínicas del International Myeloma Working Group de 2013): plasmocitoma medular que surge en hueso (vértebras, fémur y pelvis), llamado plasmocitoma solitario óseo, y otro en tejidos blandos o plasmocitoma solitario extramedular.1,2
Caso clínico Mujer de 65 años, hipertensa, que acude al Servicio de Urgencias por un episodio de epistaxis autolimitada en FND, coincidiendo con un catarro.
Se le hace una rinofibrolaringoscopia y se aprecia una lesión levemente rojiza de mucosa de aspecto normal en zona anterior de nasofaringe y rodete tubárico derecho. Se le hace una RM que muestra masa lobulada de 13 x 11 mm en mucosa anterior derecha de nasofaringe, que no se realza tras contraste ni infiltra estructuras y sin adenomegalias (Figura 1). Se informa al paciente de la necesidad de realizar una biopsia y su posible exéresis, y ésta acepta, realizándose su extirpación completa sin complicaciones mediante CENS.
El estudio histológico dio como resultado la presencia de un plasmocitoma (Figuras 2 y 3): infiltrado subepitelial por células plasmáticas con atipia que muestra fenotipo CD138+CD56+/- CD10 con restricción de cadenas ligeras kappa.
Se remite al Servicio de Hematología para completar su estudio, que solicita bioquímica, proteinograma, células plasmáticas y gammagrafía ósea que resultan normales. La cuantificación de inmunoglobulinas es normal, IF y CLLs son negativos, la punción aspirativa de médula ósea no detecta infiltración por células plasmáticas patológicas ni por citometría; y la RM cervical muestra signos de espondiloartrosis, osteocondrosis con osteofitos, y cambios degenerativos.
La paciente no presentó en ningún momento manifestaciones clínicas, y ante los resultados obtenidos, se decide someterla a revisiones periódicas. Luego de cinco años sigue asintomática, sin signos de recidiva locorregional.
Discusión
El plasmocitoma extramedular es una proliferación de células plasmáticas, considerada un linfoma de bajo grado de malignidad en la clasificación de Kiel, caracterizada por expansión monoclonal de células plasmáticas productoras de cadenas ligeras de inmunoglobulinas. Constituye el 3% de los tumores de células plasmáticas. Pueden asentarse en cualquier zona del organismo que contenga tejido linfoide (ganglios linfáticos, piel, tracto aerodigestivo), aunque se localiza preferentemente (80%) en submucosa del tracto aerodigestivo superior: en cavidad nasal (46%) senos paranasales (36%) orofaringe (25%), nasofaringe (20%), laringe (4%) y glándula tiroidea.1 Fue descrito por Schraddle en 1905, y posteriormente por Ewing. Su incidencia se estima en un caso por 500 000 personas.
Son tumores infrecuentes, que representan menos del 1% (0.4%) de las neoplasias de cabeza y cuello, y menos del 0.5% de las neoplasias malignas del tracto respiratorio superior, siendo el 4% del total de los tumores no epiteliales de cavidad nasal y senos paranasales.2
Se presenta en adultos mayores de 40 años, entre la quinta y séptima década, sobre todo en los hombres en una proporción 3:1, especialmente en los de origen caucásico. Se ha relacionado con diversos factores como irritación crónica por sustancias inhaladas e infecciones por virus, aunque no está clara su etiología. La clínica es inespecífica, dependiendo de la localización y el tamaño: tumor o edema local (80%), obstrucción nasal crónica (35%), epistaxis (35%), dolor localizado (20%), proptosis (15%), rinorrea (10%), cefalea. Raramente se presenta con disminución de la visión (parálisis del VI par craneal, proptosis, pólipo nasal, y adenopatías. Suele surgir simultáneamente en varias zonas (20%).2,3
Macroscópicamente, aparece como una masa circunscripta a submucosa de consistencia firme, no ulcerada, sésil o pedunculada, de coloración variable, sobre todo de un rojo violáceo, fácilmente sangrante. Histológicamente no se distingue del mieloma, es una proliferación difusa de plasmocitos maduros; aunque no se puede confirmar que sea una lesión precursora del mieloma.4 Al ser un tumor submucoso, deberían realizarse biopsias profundas, para que su resultado no sea negativo.
A nivel microscópico muestra una morfología blastoide con presencia de inmunoblastos, pero con inmunofenotipo de diferenciación plasmocitaria (EMA+, CD138+, CD38+, CD56+). Puede producir inmunoglobulinas, sobre todo IgG (55%) e IgA (21%). Mediante inmunohistoquímica se pone de manifiesto la producción de cadenas pesadas IgG e IgA, y cadenas ligeras kappa o lambda.5
Su diagnóstico se realiza con el estudio histológico y de extensión sistémico negativo, estudio de sangre periférica, proteinograma, proteinuria, inmunoelectroforesis sérica y urinaria, gammagrafia ósea y punción de médula ósea. Las pruebas TAC y RM son útiles para establecer la extensión de la enfermedad, careciendo de signos propios para el diagnóstico. Los criterios clínicos para su diagnóstico son: confirmación histológica, presencia de uno o dos focos de enfermedad, biopsia de médula ósea normal, radiología ósea normal, ausencia de anemia, hipercalcemia e hiperuricemia; la presencia de proteínas monoclonales en orina o suero no excluye el diagnóstico.6.7 Si se localiza en la cavidad nasal, se debe hacer diagnóstico diferencial con tumores hemorrágicos, sobre todo carcinoma escamoso, y otros como melanoma, carcinoma indiferenciado, linfoma, adenoma pituitario, entre otros.1,8
Existe controversia respecto de su evolución y tratamiento, atribuible a su baja incidencia. Generalmente, tienen una progresión locorregional: enfermedad solitaria en aproximadamente 40%, diseminación linfática del 10% al 21%, segunda localización (ósea en 38%, tejidos blandos en 20%, y ambas en un 42%, y raramente se diseminan a mieloma múltiple, incluso después de muchos años, a diferencia del plasmocitoma medular. Entre los factores que se relacionan con progresión a mieloma está se destacan la edad mayor de 60 años, el tamaño del tumor y la persistencia de proteína M luego del tratamiento.8,9
El tratamiento está cuestionado, pues se considera de elección la radioterapia, al ser altamente sensible, con una alta tasa de control local (mayor del 80%), se recomiendan utilizarla incluso después de la extirpación macroscópica total, dependiendo de la localización, a dosis de al menos 40 Gy en cuatro semanas, y si es mayor de 5 cm, dosis por encima de 50 Gy. Algunos autores aconsejan incluir todo el hueso de campo de radiación, debido a las recurrencias.10,11 Otros aconsejan la escisión de la lesión con márgenes libres por vía endonasal (CENS), siempre que el tumor sea accesible.2,9 Otros autores encuentran mejores resultados en la modalidad combinada de cirugía más radioterapia. Si los ganglios son positivos, se deben incluir en el campo a irradiar, de 40 Gy, o vaciamiento cervical seguido de RT.9-11
Conclusiones
El plasmocitoma extramedular es un linfoma de células plasmáticas de bajo grado de malignidad e infrecuente, que no afecta la médula ósea ni el tejido óseo, y aparece habitualmente en varones mayores de 40 años, como una masa de color rojo violácea no ulcerada. Puede manifestarse como epistaxis unilateral, por lo que debería realizarse una fibrolaringoscopia, como protocolo en todas las epistaxis unilaterales no recidivantes, independientemente de la edad, para descartar tumores a nivel de la rinofaringe.
1. Gerry D, Lentsch EJ. Epidemiologic evidence of superior outcomes for extramedullary plasmocityma of the head and neck. Int Arch Otorhinolaryngol 17(2):213-217, 2013. doi: 10.7162/S1809-97772013000200016.
2. Tisner JV, Fraile J, Ortiz García A. Plasmocitomas primarios extramedulares de vías respiratorias altas. Estudio de cuatro casos de localización reciente. ORL Aragón 0:6-9, 1997.
3. Garcia C, Armengot M, Sabater V, Reboll RM, Frias S, Basterra J et al. Plasmocitoma solitario extramedular de amígdala tubárica. Acta Otorrinolaringol Esp 60(4):301-3, 2009.
4. Bachar G, Goldstein D, Brown D, Tsang R, Lockwood G, Pérez-Ordóñez B, et al. Solitary extramedullary plasmacytoma of the head and neck long-term outmcome analysis of 68 cases. Head and neck 112-9, 2008. DOI: 10.1002/hed.20821.
5. Creach KM, Foote RL, Neben-Wittich MA, Kyle RA. Radiotherapy for extramedulary plasmacytoma of the head and neck. Int J Radiation Oncology Oncology Biol Phys 73(3):789-94, 2009. Doi:10.1016/j.ijrobp.2008.04.077.
6. Pont E, Mazón M, Del Campo J, Viel M. Persistent plasma cell tumor of the nasopharynx after radiotherapy treatment: a case report and review of the literature. J Otol Rhinol 3(6), 2014.
7. Dos Anjos MA, Granato L, Ikeda F, De Própero JD. Extramedullary nasal plasmacytoma: Literature review and a rare case report. Otolaryngol Head Neck Surg 148(6):974-81, 2013.
8. Thumallapally N, Meshref A, Mousa M, Terjanian T. Solitary plasmacytoma: population-based analysis of survival trends and effect of various treatment modalities in the USA. BMC Cancer 17:13, 2017. doi: 10.1186/s12885-016-3015-5.
9. Ozsahin M, Tsang R, Poortmans P, Belkacemi Y, Bolla M, Dinçbas FÖ, et al. Strojan P, Soba E, Lamovec J, Munda A. Extramedullary plasmacytoma: clínical and histopathologic study. Int J Radiation Oncology Biol Phys 53(3):692-701, 2002.
10. Outcomes and patterns of failure in solitary plasmacytoma: a multicenter rare cancer network study of 258 patients. Int J Radiation Oncology Biol Phys 64(1):210-17, 2006. Doi: 10.1016/j.ijrobp.2005.06.039.
11. Sasaki R, Yasuda K, Abe E, Uchida N, Kawashima M, Uno T, et al. Multi-institutional analysis of solitary extramedullary plasmacytoma of the head and neck treated with curative radiotherapy. Int J Radiat Oncol Biol Phys 82(2):626-34, 2012. doi: 10.1016/j.ijrobp.2010.11.037.
Está expresamente prohibida la redistribución y la redifusión de todo o parte de los contenidos de la Sociedad Iberoamericana de Información Científica (SIIC) S.A. sin previo y expreso consentimiento de SIIC.
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου